- To review embryology of WD. - To discuss pathologies related to WD anomalies and their typical association.
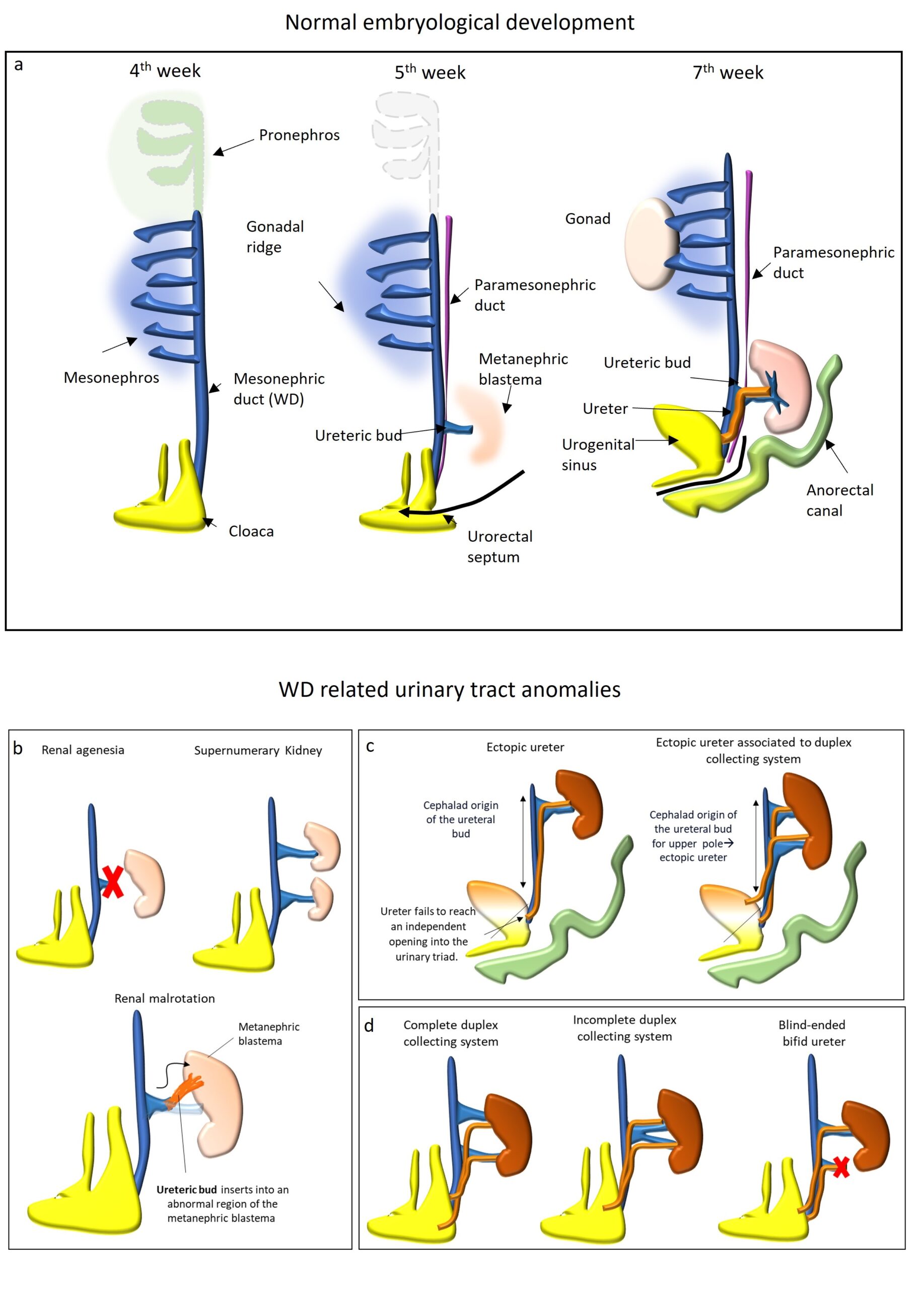
WD develops in the early stages of embryonic development, and it has a pivotal role in the genitourinary system formation. In general, 1/3 of congenital anomalies are located within the kidneys or the urinary tract and the majority do not cause clinical problems [1]. The mesonephric duct took its name from Caspar Friedrich Wolff, a German physiologist and embryologist who firstly described it in 1759 [2]. Although malformations of the genitourinary tract usually are diagnosed during childhood, they can remain silent and be incidentally detected during the evaluation and/or treatment of other pathologies in adulthood. The main purpose of our work is to review WD embryology and anatomy to offer a tool to Radiologists for better understand and diagnose WD related pathology and associated congenital malformations. Through a multi modal approach, Radiologists will familiarize with these conditions and guarantee a correct diagnosis not only in paediatric patients but also in the adult population. Embriology Between 3rd and 8th weeks of gestation, the urogenital system develops from the intermediate mesoderm, which extends along each side of the dorsal body cavity of the embryo and forms the urogenital ridge (during the 4th week). The urogenital ridge giving rise to the nephrogenic cord and three successive kidneys form over a few weeks (pronephros, mesonephros, and metanephros). At the beginning of the 4th week, at the cervical region, the pronephros (a non-functional primitive kidney) develops but it totally regresses by day 25. At the end of the 4th week, the intermediate mesoderm generates the mesonephros in the thoracolumbar region. The mesonephros is a functional primitive kidney with excretory tubules which drain into the WD for a brief period [3], Fig. 1. Indeed, the WDs, originate as excretory ducts and appear near the gonadal precursor of both sexes shortly after their formation. Soon after, a second pair of ducts called paramesonephric or Müllerian ducts (MD) form laterally to WD. During the 5th week, the WD connects the primitive kidney to the cloaca. The cloaca is a common chamber into digestive, urinary, and reproductive tracts discharge their contents. A cloaca exists in all human embryos up to 4–6 weeks, at which time it becomes partitioned into rectum and urogenital sinus. At the 5th week of development, the ureteric bud arises as a diverticulum from WD, Fig. 1. The ureteric bud is a pivotal structure that differentiate into the ureter and the collecting duct system of the kidney. The ureteric bud grows laterally and invades the center of the metanephric blastema, the primordial renal tissue [4]. The meeting of these two tissues causes changes in the bud and in the metanephros. The ureteric bud divides and branches forming the collecting system (renal pelvis, infundibulae, calyces, and tubules) for urine drainage. The metanephric blastema forms glomeruli, proximal and distal tubules. This process is known as “induction of the kidney”. The distal portions of the mesonephric ducts and attached ureteric buds descends inferiorly to meet the urogenital sinus and become incorporated into the posterior wall of the primitive bladder. At 9th weeks of development, the metanephros (mature kidney) starts to produce urine and the caudal end of WD is called common excretory duct. Progressive incorporation of the common excretory duct into urogenital sinus leads to separate openings of the ureter and WD: the ureter empties into the urogenital sinus cephalad to the WD. In this way, the urogenital sinus is divided by the orifices into two different portions: the cephalad one that will become the bladder and caudal one that will become the urethra. In females, WDs regress and the ureter remains alone. In males, the establishment of internal reproductive system involves two key events: the formation of the testis and the maintenance and differentiation of the WD. Although not essential for initial WD formation, the later differentiation of this embryological structure relies on androgens derived from the Leydig cells. In the presence of a Y chromosome and the SRY gene, müllerian‐inhibiting substance produced by Sertoli cells causes regression of the MD. Meanwhile, testosterone produced by the Leydig cells induces differentiation of the WD into the efferent ducts that communicate with the rete testis, followed by the epididymis, ductus deferens, seminal vesicle, and ejaculatory duct. Anomalies related to proximal WD anomalies Congenital Anomalies of kidney: The WD is central in the development of the urogenital system and its role is well-established. The kidney develops because of cross-talk between the WD (later, the ureteric bud) and the future metanephric blastema. Each step of renal development is dependent on the expression of different genes and their protein products that stimulates the so-called induction of the kidney. The general classification of anomalies of the upper urinary tract includes renal anomalies of form, position, and number and anomalies in the development of the urinary collecting system. A complete review of the kidney congenital anomaly is beyond the scope of this paper and the reader is referred to the literature for further details [3]. The aim of our is to discuss only anomalies related to WD anomalies: renal number anomalies (agenesia or supernumerary kidney), renal malrotation and WD related collecting system anomalies, Fig. 1. Renal agenesia (RA) is due to a defect of the WD, the ureteric bud, or the metanephric blastema. It occurs because of failure of the cross-talk between ureteric bud and metanephric blastema [5]. RA can be bilateral (1 /4000 births), with a predominance in males and it is fatal [3]. Affected infants can develop oligohydramnios and the so-called Potter Sequence with immature lungs and Potter facies (hypertelorism, prominent inner canthal folds, and recessive chin) [6]. Unilateral RA is often asymptomatic with an estimated incidence 1/ 450 to 1000 births. It can be discovered incidentally during imaging examinations performed for other reasons. It is often associated with other anomalies in the contralateral kidney UPJO (3%), vesicoureteral reflux (28%), obstructive megauretere (11%) or other organs (eg, cardiac, genital, or gastrointestinal organs) [5]. Genital anomalies are also frequently associated with RA (see below) and for this reason, in case of unilateral RA, Radiologist must always check WD derived organs anomalies in male (especially seminal vesicle) and evaluate uterine morphology. Supernumerary Kidney (SK) is an additional accessory organ most located caudally to the left kidney. SK on the right side or bilateral SKs rare (less than 100 cases reported in the literature) [7]. Presence of two independent ureteral buds, arising separately from the WD, penetrating the metanephric blastema, and dividing in two. SK is usually asymptomatic. Urinary complications may occur, such as urinary incontinence, pyelonephritis, pyonephrosis, renal and ureteral calculi, and malignancy. Also, SK can be associated with other congenital anomalies (eg, vaginal atresia, duplication of the penis or the female urethra, coarctation of the aorta, and other abnormalities) [8]. On imaging, the main differential diagnosis of the SK includes evaluation for a duplex collecting system. The SK is typically smaller than the native kidney and it may be completely separated from the ipsilateral kidney or fused by parenchymal or fibrous tissue. SK has its own arterial supply from the aorta or the common iliac artery, and drains via the inferior vena cava, with a distinct collecting system and an encapsulated parenchyma. Renal malrotation is classified as a malposition renal anomaly and can be considered as a WD related anomalies. Renal malrotation can be unilateral or bilateral (M>F), prevalence 1/200 and is usually related to other anomalies. It is defined as an abnormal position of the kidneys in relation to the hilum due to an anomaly of ureteric bud insertion into an abnormal region of the metanephric blastema. Because of its association with renal ectopia, the process of ascent and rotation are probably related [9]. Renal malrotation is often asymptomatic and the diagnosis is usually made incidentally. Congenital Anomalies of the collecting system Congenital Anomalies of the collecting system related to WD anomalies are duplex collecting system, primary vesico-ureteric reflux ectopic and ureter, Fig. 1. Duplex collecting system is the most common congenital ureteral abnormality. It can be complete or incomplete. Embryological causes are summarized in figure 1. The ureteric bud can divide into 2 branches. If the additional branch reaches the metanephric blastema, this leads to complete duplication. If the additional branch fails to reach the metanephric blastema, a blind-ending ureter or incomplete duplication develop [10]. Duplex collecting systems are usually asymptomatic, incidentally diagnosed with no associated renal abnormalities, and are considered a normal variant. When renal abnormalities are present, they tend to affect the whole kidney in incomplete duplications and can affect either one or both poles in complete duplications. In complete duplex collecting systems, the lower pole follows the normal anatomic structure, with an orthotopic ureter implantation, while the ureter of the upper pole has an ectopic bladder implantation, inferiorly and medially displaced, and, therefore, is prone to be associated with other abnormalities: embryology explains the mnemonic rule “upper pole obstructs, lower pole refluxes”. A blind-ending bifid ureter is considered an uncommon variant of duplex collecting system anomalies and it is more common in women and in the right site [11]. Blind-ending bifid ureters can be classified into subtypes depending on the origin of the branch, respectively proximal, middle (least common), or distal. Blind-ending bifid ureters are typically asymptomatic. Symptoms may include abdominal pain and recurrent urinary tract infections. Most cases are detected incidentally during common urological investigations such as intravenous urography, retrograde pyelography, or diagnostic cystoscopy, CT and MRI. Especially CT and MRI allow a detailed evaluation of urinary system anatomy and exclusion of other possible diagnosis, Fig. 2. Primary vesico-ureteric reflux (VUR) VUR is defined as abnormal flow of urine from the bladder into the upper urinary tract. In the developing embryo, if the ureteric bud develops more caudal along the WD, its final location within the bladder will be abnormally cranial to the normal site. This results in poor development of a “short” ureterovesical tract and VUR. The International Reflux Grading System (IRGS) divides RVU in five grades: (I.) Reflux only into the ureter; (II.) Reflux into the entire ureter and pelvicalyceal system, without dilatation; (III.) Mild pelvic or ureteral dilatation, with mild or no blunting of the fornices; (IV.) Moderate dilatation of the pelvis and kinking of the dilated ureter, with moderate dilation of the calyces; (V.) Massive ureteral and/or pelvicalyceal dilatation. The maximal degree of VUR is used for grading and should be accurately described; intrarenal reflux should be reported separately because intrarenal reflux may predispose to developing renal failure. Reflux predisposes to renal infection (pyelonephritis) and kidney scar formation. Reflux nephropathy is a common cause of hydronephrosis and renal failure; therefore, it is important a prompt diagnosis and treatment. US well depictes and grades hydronephrosis. The diagnostic procedure for evaluation of VUR, especially in males is VCUG; Echo-enhanced cystosonography has recently been proposed as a promising new method for detecting and grading VUR without exposing patients to ionizing radiation. [12] Ectopic ureters If ureter inserts into other position than the superolateral portion of the trigone is defined as ectopic. Ectopic ureters are more common in females than in males (F.M= 5:1). Possible sites of ectopic exit are posterolateral wall of the urethra, anterolateral vagina, vestibule and rarely the cervix and uterus in female and in the seminal vesicles in male. Ureteric ectopia results from abnormal development of the WD and MD systems at 5 to 8 weeks of gestation [13]: If the ureteral bud develops more cephalad on the WD, it will not achieve an independent opening into the urinary triad. The ureter remains attached to the distal part of the WD leading to a more caudal insertion. Abnormal development of the ureter can cause maldevelopment or absence of the ipsilateral kidney. Often, a rudimentary renal unit is detected on the involved side and its ureter ends in the ipsilateral vas deferens or seminal vesicle. As mentioned before, complete duplex collecting system is often associated to ureter ectopia of ureter draining the upper renal pole due to its cephalad origin from WD. Enlarged ectopic ureters are normally diagnosed by ultrasound for work-up of pyuria. Several imaging techniques have been used in the evaluation and differentiation of pelvic anomalies: US is the first level imaging technique, but MRI can offer a more detailed anatomical information, Fig. 2. Congenital and benign pathology related to WD anomalies in male: In male, congenital malformations related to WD anomalies consist of anomalies of epididymis, vas deferens, seminal vesicles, and ejaculatory ducts. All the WD deriving structures have an ample variety of congenital anomalies (isolated or in combination). Congenital anomalies of structures derivated from WD can be classified as abnormalities of number (agenesis, duplication, fusion), maturation (hypoplasia) and canalization (cysts) [14]. On imaging, agenesis is characterised by non-visualisation of the anatomical corresponding structure (e.g. seminal vesicles). Among WD anomalies, SV cysts are the most common and can be congenital or acquired. Accumulation of secretions in the gland resulting from insufficient drainage, due to atresia of the ejaculatory ducts, causes distention, leading to formation of a cyst that develop during the years of sexual activity and may become symptomatic in the second to third decade. SV cysts less than 5 cm may remain asymptomatic or can manifest with infective or irritative urinary symptoms. Large or giant cysts (> 12cm) may cause obstruction of the bladder or bowel. Moreover, they can be associated to chronic recurrent prostatitis and epididymitis, painful ejaculation, hematuria, perineal/ scrotal pain, infertility, or hematospermia. SV cysts are located within SV, posteriorly to the urinary bladder, superiorly to the prostate gland. On US, SV cyst appears as an anechoic mass in the SV. US is also useful for guide needle aspiration of the lesion. On CT, SV cyst may appear as a low attenuated lesion. On MR, it appears as a unilocular round or oval cystic lesion with hyperintense signal intensity on T2-weighted images (fluid content) and hypointense signal on T1-weighted images (increased T1 signal intensity can be secondary to previous infection or bleeding). There may be contrast enhancement only of the walls of the cyst. Transrectal ultrasound (TRUS) is considered the modality of choice for the initial assessment of the seminal vesicle cysts in adulthood otherwise for adolescent patients, it is preferable to use firstly transabdominal ultrasound [14]–[16]. The proper diagnosis of prostatic and periprostatic cyst can be obtained firstly defining its location (intra- versus extra-prostatic cyst). Then, in case of intraprostatic lesion, the second step is to define its position (median versus paramedian versus lateral ones) [16]. Pelvic cysts are non-touch lesions. While in case of symptoms, surgical excision should be preferred to aspiration, due to its association with high risk of recurrence. Cysts of the epididymis are the most common epididymal mass and they are diagnosed in 20-40% of asymptomatic patients; they can be multiple and bilateral. These masses may be either true epididymal cysts, which are lined with epithelium, contain clear serous fluid (not contain sperm) and occur anywhere within the epididymis or they may be spermatoceles which originate from obstruction and dilatation of the efferent ductal system and are filled with sperm lymphocytes, and cellular debris and generally occurs in the head of epididymis. The pathogenesis of epididymal cyst is not certain, Nistal and al. described a new congenital epididymal lesion in association with renal and urinary tract malformations, called cystic dysplasia of the epididymis. The simultaneous occurrence of these anomalies may be related to disrupted WD development [17]. On US scans, the cysts are usually an anechoic, well-defined lesion, but larger cysts may have internal septa and filled with corpuscular material, that appears mobile and hyper-reflective at Color Doppler. The cyst cannot be distinguished sonographically from a spermatocele. Asymptomatic cyst not required treatment (only clinical and US follow up), but if they become symptomatic or start increasing in size, surgical removal or sclerotherapy should be taken into consideration [18], [19]. Seminal tract obstruction can be classified according to the level of obstruction, into proximal seminal tract obstruction (epididymis and scrotal portions of the vas deferens) and distal seminal tract obstruction (inguinal, pelvic and ampullary portions of the vas deferens, and ejaculatory ducts) [20]. Ejaculatory duct obstruction (EDO) should be suspected in male patients with azoospermia or severe oligozoospermia, low ejaculate volume (<1.5mL), low semen pH, no fructose, and normal secondary sex characteristics, testes, and hormonal profiles, especially in association with dilatated SV. Seminal tract obstruction can be congenital (due to ductal atresia or agenesis, stenosis, mutations of CFTR-cystic fibrosis transmembrane regulator- gene and ectopic ureteral orifice opening into the ejaculatory duct) or acquired (due to infection/inflammation, trauma, pelvic surgery, prolonged catheterization and stone formation in the distal duct). It can be associated with other WD anomalies (ipsilateral renal agenesis and SV cyst) in the Zinner syndrome (see below). Patients can be completely asymptomatic or may present painful ejaculation, perineal pain, hematospermia, dysuria, or difficulty with defecation. The diagnosis requires integration of clinical data, laboratory tests, semen analysis, and imaging exams. When physical examination, laboratory tests and semen analysis results are normal a post testicular (obstructive) cause of azoospermia should be suspect (up to 40% of cases). The imaging work-up of patient affected by azoospermia is firstly to perform scrotal and Transrectal US (TRUS) [21]. MRI is considered a second level imaging technique considered superior to TRUS in delineating the anatomy of seminal tract and could offer a “detailed map” for guiding interventional diagnostic or corrective procedures. The gold standard treatment is Transurethral Resection of Ejaculatory Duct but there are other options such as Transurethral Vesiculoscopy, Balloon Dilation and Cyst Aspiration. Since spermatogenesis is preserved in men with EDO, another valid alternative for couples that want a pregnancy is to use Assisted reproductive technology [20], [22]. Congenital and benign pathology related to WD anomalies in female: Small remnants of WD can extend from the mesosalpinx via the broad ligament to the cervix. From cranial to caudal, these remnants are defined as epoophoron if they lie between fallopian tube and ovary, paroophoron if they lie below the ovary in the broad ligament, and Gartner's ducts and cysts along the cervix and vagina [23]. POC appears as homogeneous, well-defined lesion with high signal intensity on T2-w images located in proximity of the ovarian parenchyma. GDCs are visualized as a cystic lesion usually located in the antero-lateral wall of the upper vagina, above the most inferior aspect of the pubic symphysis WD remnants, due to epithelial secretory potential, can be the origin of benign pathologies such Gartner's duct cysts, mesonephric hyperplasia (especially histological finding), paraovarian /paroophron cysts. Gartner duct cysts (GDCs) originate from vestigial remnants of WD and consist of lined with non-mucinous bland, cuboidal or low columnar epithelium [23]. The majority are solitary, less than 2 cm and asymptomatic but they can be larger and symptomatic (for example urinary tract symptoms if they cause mass effect on the urethra) and imaging studies become necessary to make a differential diagnosis. On imaging, GDCs are visualized as a cystic lesion usually located in the antero-lateral wall of the upper vagina, above the most inferior aspect of the pubic symphysis [24]. On MRI, signal intensity on T1-w can be intermediate to high signal intensity due to proteinaceous or haemorrhagic components, Fig. 5 [25]. Radiologically, differential diagnosis with other vaginal/paravaginal cysts is based on anatomic landmarks (position respect the pubic symphysis) and location respect the vaginal wall. GCDs may be associated with renal and ureteral anomalies, such as ectopic ureter, URA, and hypoplasia [25]. Although these cystic lesions are generally asymptomatic and not associated with increased risk for malignancy, conservative treatment with cyst aspiration and tetracycline sclerotherapy, or surgical excision may be used to treat larger and symptomatic lesions [23]. Paraovarian/paroophron cysts are defined as cysts that arise from the broad ligament (mesosalpinx) between the fallopian tube and the ovary, represent about 10%–20% of all adnexal masses [26]. Usually POCs are solitary, asymptomatic small lesion (the average size is 8 cm) but they may become symptomatic when they enlarge (under hormonal influence) or in case of haemorrhage, rupture or torsion [26]. Preoperative diagnosis of POC with imaging can be difficult. On Imaging, they are usually visualized as a simple, round or oval, unilocular cystic lesion. Different radiological signs are useful to differentiate POC from ovarian lesion (“Split sign”, embedded sign, beak sign). On MR imaging POC usually appears as homogeneous, well-defined lesion with high signal intensity on T2-w images and low signal intensity on T1-w images (high signal intensity on T1-w images and thicker walls in case of torsion or haemorrhage), Fig. 5. The presence of associated solid tissue is indicative of a neoplasm [26]. Since there aren’t certain criteria for treatment, PCOs are usually management as ovarian lesion. Complex malformation syndrome Male: Zinner syndrome Zinner’s syndrome (ZS) is a rare condition characterized by the triad of unilateral RA, ipsilateral SV cysts, and ipsilateral EDO. This association is causes by an abnormal development of the WD between the 4th and 13th week of gestational age: the ureteric bud fails to join the metanephros blastema which leads to renal agenesis and atresia of the ipsilateral ejaculatory duct. The gonad continues to develop, and insufficient drainage of the seminal fluid results in the cystic structure of the SV. ZS is quite rare and asymptomatic. Therefore, patients are usually diagnosed during 2nd to 3rd decade of life and they typically present urinary symptoms (such as pollakiuria, dysuria, increased urinary frequency, haematuria) perineal and ejaculatory pain, or epididymitis. Due to EDO, approximately 45% of patients are affected by infertility. Imaging show a periprostatic cystic mass and ipsilateral RA, Fig. 3. For the appearance of SV cyst on imaging and its differential diagnosis, see above. In addition, other anomalies such as an ectopic ureteral insertion into the SV, ejaculatory duct, prostatic urethra, or vas deferens or agenesis of the vas deferens may be associated. For asymptomatic patients, follow up is acceptable. Surgical treatment is reserved for symptomatic patients (from transurethral unroofing to open surgery with vesiculectomy with or without vasoligation) [3], [27], [28]. Female complex malformative syndrome Among female uro-genital tract malformations, obstructed hemivagina and ipsilateral renal agenesis (OHVIRA) syndrome, also known as Herlyn-Werner-Wunderlich syndrome, is a rare syndrome thought to be related to WD abnormality. It is characterized by the classic triad of uterus didelphys, obstructed hemivagina, and ipsilateral RA [29]. The pathogenesis of OHVIRA syndrome is controversial and thought to be related to an anomaly in development of both MD and WDs but WD seems to have a primary role. Indeed, as we have seen before, the ureteral bud sprouts from WD and its absence or distal injury will give rise to RA or other urinary tract malformations (dysplastic or polycystic kidney, ectopic or duplicated ureters) and explained their presence the syndrome. WD has also an important role in the female genital system development: WD, acting as a guide element, induces the fusion and resorption of MDs. Fusion failure of the MDs leads to uterine duplicity (didelphys and bicollis). Vaginal abnormality origins are controversial because, according to the classic theory, the upper two-thirds of the vagina originate from MD while the inferior third from the urogenital sinus. However, this theory couldn’t completely explain the presence of obstructed hemivagina in the OVHIRA syndrome. Some authors described the WD participation in the development of the vagina. Ancién proposed a different hypothesis where the vagina seems to arise completely from the fused WD and highlight its fundamental role for the adequate formation and cavitation of the vagina (Acién, 1992) [30]. It is important to note that ovaries a have a different embryological origin (genital ridge) and this explain way usually are not involved in MD anomalies. OHVIRA is typically diagnosed during the peri-pubertal period. The vaginal septum can obstruct the menstrual flow leading to lower abdominal, progressive dysmenorrhea, or cystic masses in the vaginal wall after menarche. Retrograde flux predisposes to endometriosis and pelvic infections (abscesses, pyosalpinx, and peritonitis. Infertility or miscarriages due to endometriosis and adhesions can occur. Coexisting urologic anomalies can result in recurrent urinary tract infections [29]. Transvaginal US detects uterine anomalies (didelphic/bicornuate uterus), with or without hematometra and hematocolpos. Abdominal US can identify ipsilateral renal agenesis. MRI is considered the gold standard for imaging and identifying Müllerian anomalies, as it provides details about uterine and, especially, vaginal morphology (localization and thickness of the vaginal septum) and information about the presence of communication between the two cervices or vagina. MRI can easily detect hematocolpos, hematometra and hemoperitoneum due to the high signal intensity in T1w images [31]. Hysterosalpingography is only of interest for the detection of a communication between the 2 uterine cavities. There are a lot of classifications of malformations of the female genital tract, such as the ESHRE/ESGE classification system, based on the uterine anatomy/morphology and with independent co-existent sub-classes for cervical and vaginal anomalies [32]. These classifications tend to simplify the approach to genital malformation, and they are very useful in the clinical practice but with the risk to lose etiopathogenetic knowledges that are essential to rise the suspicious of associated anomalies and to guide an appropriated diagnostic and therapeutic iter. For these reasons, Ancién et Al. proposed an embryological-clinical classification that is very useful to understand these malformations and what are the embryological structure involved and, consequently, the possible associated malformations that radiologists and clinics must search or rule out. According to the embryological-clinical classification for female genitourinary malformations proposed by Acién et Al., female genital tract malformation can be due to: Agenesis or hypoplasia of an entire urogenital ridge that occurs early during embryogenesis and can have Müllerian agenesis (aplasia of the uterus and upper part of the vagina) known as Rokitansky syndrome type II or unicornuate uterus. Both forms are associated functioning ovary (due to different origins), and renal anomalies (especially agenesia, e.g. controlateral to the unicornuate uterus). This type can also be associated with vertebral, cardiac and auditory anomalies. Mesonephric anomalies with an absence of the WD opening into the urogenital sinus and of the ureteral bud sprouting. These are indeed recognized as a specific subgroup of malformations where renal agenesis and ipsilateral blind vagina and uterine anomaly coexists. Isolated Müllerian anomalies can affect the MD (with tubarian and uterine malformations), the Müllerian tubercle (with complete vaginal agenesis or atresia and segmentary atresias as transverse vaginal septum) or both Müllerian tubercle and ducts (Rokitansky or MRKH syndrome type 1) with no renal agenesia. This classification also includes anomalies due to Gubernaculum or urogenital sinus dysfunctions (like imperforated hymen or congenital vesicovaginal fistulas) and other malformative combinations (WD, MD and/or associated cloacae anomalies) [30]. According to the retrospective study of Zhu et al, mesonephric anomalies are classified into two classes. This classification is important to understand clinical features and management. Class 1: complete obstruction of the hemivagina characterized by hematocolpos, hematometra, hematosalpinx and possible hemoperitoneum (and secondary endometriosis), Fig. 4. Early age and acute onset with abdominal pain, fever, and vomiting. Divided into: - class1.1 blind hemivagina (no communication between the duplicated uterus and vagina); - class 1.2 cervicovaginal atresia without communicating uterine (menses from the uterus behind the septum cannot outflow through the atretic cervix). Class 2: incompletely obstructed hemivagina, divided into: - class 2.1 partial reabsorption of the vaginal septum (small communication between the two vaginas), Fig. 4. - class 2.2 with communicating uteri (small communication between the two cervices), Fig. 4. In this class menses can outflow through the small communication, but the drainage is impeded. The age of onset is later with purulent or bloody chronic vaginal discharge and possible ascending genital system infection [31]. In OVHIRA syndrome, on the affected side (homolateral to RA), dilated tortuous blind-ended ureter, ureterocele or other tubulo-cystic malformation of WD remnants can be detected, Fig. 4 [33]. The final objective of optimal surgery for patient affected by OVHIRA syndrome is to relieve symptoms and retain fertility through a wide resection of the vaginal septum and/or laparoscopic resection of the atresic uterus (for class 1.2) [31]. Female malignant pathology related to WD: Mesonephric adenocarcinoma Mesonephric adenocarcinoma (MA) is an extremely rare neoplasm of female genital tract that arise from mesonephric WD remnants, most commonly in the uterine cervix (lateral wall), rarely in the vagina. Mesonephric-like adenocarcinoma (MLA) is a recently new group of probable mesonephric adenocarcinomas of the ovary and uterine corpus, but their histogenesis is not firmly established [34]. The incidence of MA is uncertain because it is frequently confused with other adenocarcinomas or interpreted as benign florid mesonephric hyperplasia, but it is reported as <1% of all cervical adenocarcinomas and only 23 cases of vaginal MA have been escribed [35], [36]. This neoplasm occurs over a wide age range, but infrequently is diagnosed in patients under 30 years of age. The most common clinical presentation is abdominal pain or pelvic pain, but also abnormal bleeding and vaginal discomfort, with or without a clear mass on physical exam [23], [37]. Due to the rarity of this disease entity, little is known about its imaging features. At MRI, it is described as a pelvic mass that arise from cervix, from vagina Fig. 5, from uterine corpus and ovary with or without extension to the adjacent organs [36], [38]. Even at transvaginal ultrasound, MA has been described as a pelvic mass, hypoechoic or cystic [38]. It must be differentiated from other malignant pathologies, such as well differentiated villoglandular adenocarcinoma of cervix, adenoma malignum, clear cell or endometroid adenocarcinomas, female adnexal tumor of probable Wolffian origin and metastasis, depending on its location [39]. Pathological diagnosis is also challenging since typically exhibits a mixture of morphologic patterns, including tubular, solid, papillary, retiform, ductal and malignant spindle cell biphasic pattern. MA is commonly positive with CD10, vimentin, EMA, calretinin, and inhibin, whereas ER, PR and CEA are usually negative. PAX8, HMGA2, and CA125 are usually positive, and TTF-1 and HNF1-b can be expressed. WT1 is negative. KRAS and NRAS are commonly mutated [23]. Prognosis cannot be precisely predicted due to the small number of cases; most mesonephric neoplasms presented at an advanced stage (II–IV) (60% MA of the cervix), and 52% developed recurrences. Distant metastases at initial diagnosis were found in <5% of patient, but a malignant clinical course has been reported in about 40% of patients [34], [37]. Owing to the small number of cases, there is no consensus about MA treatment and it depends on its stage. In early stage, the first line treatment is radical surgery (hysterectomy with or without bilateral salpingo-oopherectomy and pelvic lymph node dissection). Usually first-line adjuvant chemotherapy protocol after surgery is 4-8 cycles of carboplatin and paclitaxel. The therapy for recurrent disease included gemcitabine, carboplatin, and paclitaxel [37], [38].
Figure 1 Box a Schematic presentation of embryological development of urinary tract; Box b WD related kidney anomalies; Box c and d WD related collecting system anomalies.
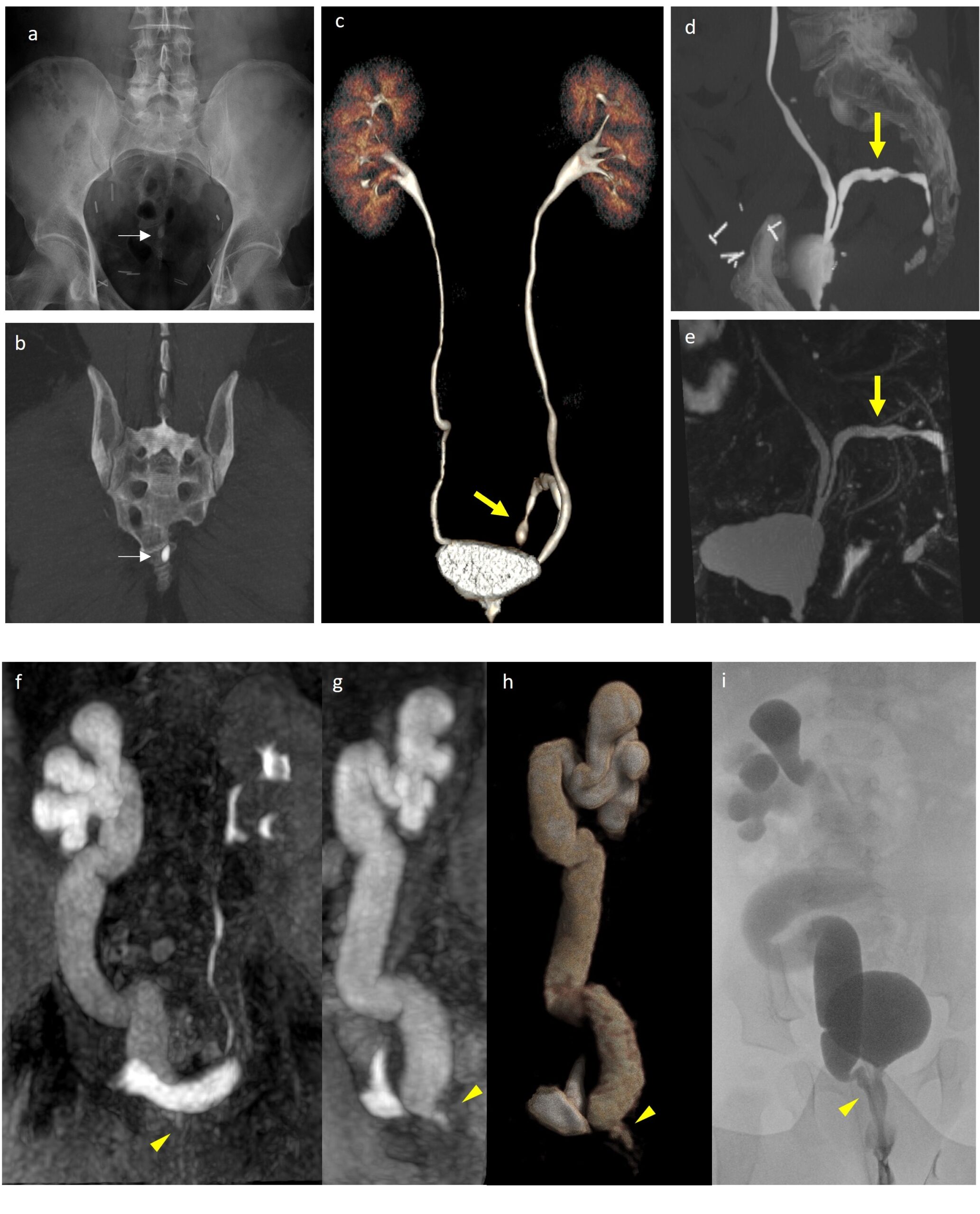
Figure 2 Blind-ended left ureter incidentally discovered during CT urography for a suspect leakage at vescico-urethral anastomosis after prostatectomy. Abdominal Xray (a) and before contrast CT (b) shows some pre-sacral calcification due to lithiasis of the blind-ended ureter (arrow). CT Volume rendering Reconstruction (c), CT MIP image (d) and 3d static fluid MR urography (e) allow a detailed evaluation of urinary system anatomy and exclusion of other possible diagnosis. Ectopic vescicoureteral junction. 10-month baby underwent MR due to pelvis enlargement (>10 mm), diagnosed during pre-natal US. SPIR T1-WI MIP reconstruction on coronal (f) and sagital planes (g) and 3D reconstruction (h) show homogeneus right ureter enlargment and ectopic vescicoureteral junction into proximal tract of urethra (arrowhead). Voiding cystourethrogram (VCUG) (i) shows V grade vescicoureteral reflux during urination due to stenotic and insufficiency in ectopic vescicoureteral junction.
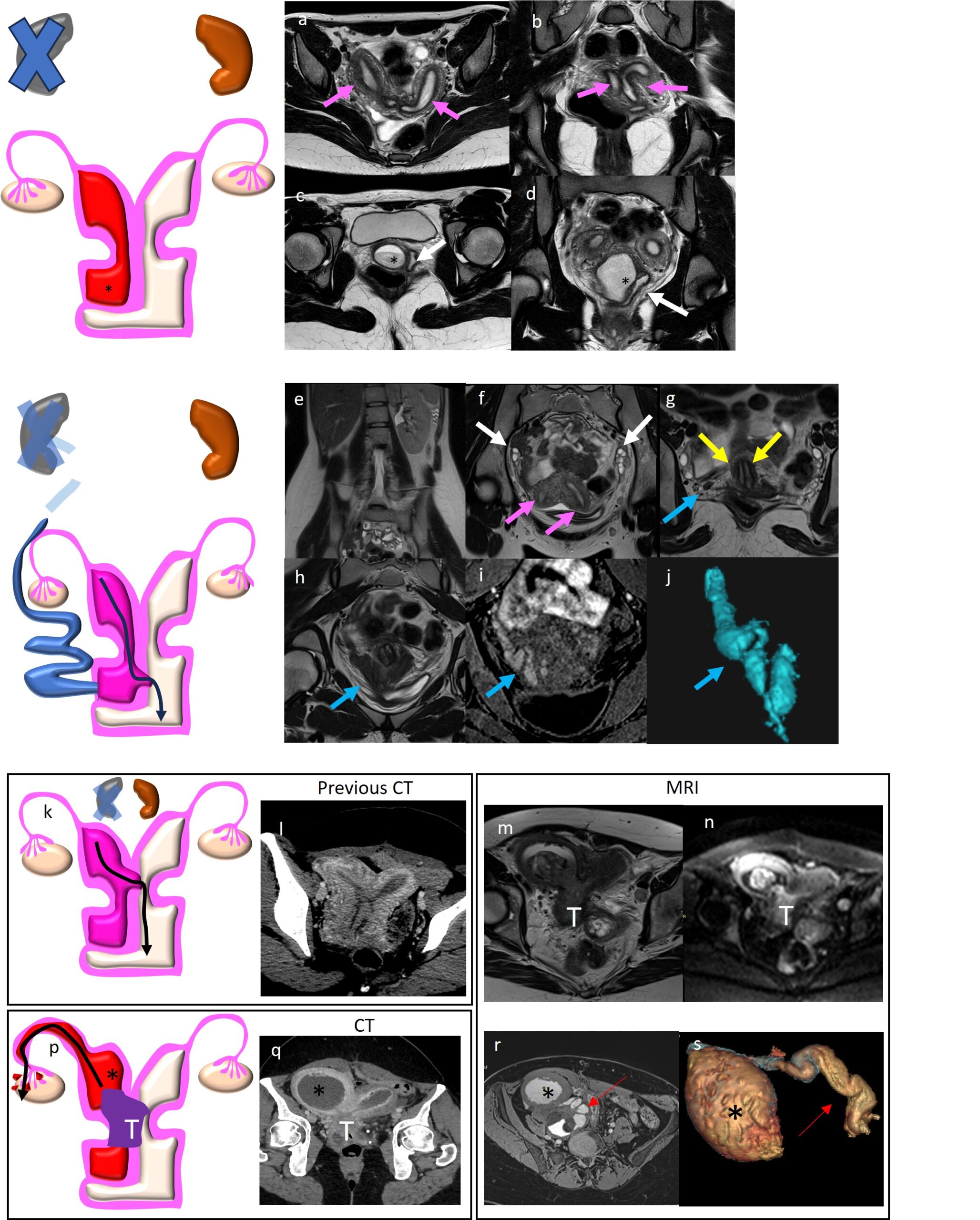
Figure 3 Zinner Syndrome. Follow-up US in a 14 years old boy with known left renal agenesis (a) showed enlarged cystic forrmation (arrowhead) next to the bladder on the posterior left side ( b). Coronal fat-sat T2-WI (c), axial T2-WI (d), SPIR T1-WI axial MIP (e) and 3D reconstruction (f) better depict the cystic formation, referable to seminal vescicle cyst.
Figure 4 A,B,C,D) OHVIRA Syndrome with complete right hemivagina obstruction (class 1) characterized by right hematocolpos (* in c and d), bicornuate (arrow in a) and bicollis uterus (arrows in b). Canalized left hemivagina (white arrow in d). Right RA not shown. E, F, G, H, I, J) OHVIRA Syndrome with incompletely obstructed hemivagina characterized by partial reabsorption of the vaginal septum associated to bicornuate and bicollis uterus (pink arrow in f and yellow in g), right RA (in e). Coronal T2-WI (h), coronal fat sat T1-WI (i) and 3D reconstruction (j) showed also right tubulo-cystic remnant of WD (blue arrow also in g). OHVIRA class 2.2 with communicating uteri (k, l) who developed cervical tumor (T in p, q, m, n) with consequent right hematometra (* in r, s) and right hematosalpinx (red arrow in r,s) better demonstrated by 3d reconstruction (s).
Figure 5 WD related benign pathologies arise from small remnants of WD can extend from the mesosalpinx via the broad ligament to the cervix. T2WI (a,b) showed simple paraovarian cysts. Gartner's ducts cyst (dashed arrow in c,d,e) is located in the antero-lateral wall of the upper vagina (better depicted on axial T2wi in e), above the most inferior aspect of the pubic symphysis (sagittal T2WI in d). Mesonephric adenocarcinoma in a 50 years old patient with a previous history of hysterectomy due to endometriosis. MRI shows a solid mass with a cystic component in proximity of the vaginal vault (red arrow) on sagittal (f) and axial (g) T2WI. 3D static fluid MRI urography (h) showed bilateral hydronephrosis due to bilateral pathological iliac lymph nodes (white arrow in I and j) with signal restriction on DWI (j). Fusion imaging between axial T2WI and DWI is showed in k.
The aim of our work is to review embryology, anatomy and pathologies related to WD through a multimodality approach to make radiologists more aware of such conditions and their typical associations and to guarantee a correct diagnosis not only in paediatric patients but also in the adult population. References [1] M. Cooper, C. Guiterrez, H. Swana, M. Rich, and L. Wiegand, ‘Images–A series of congenital mesonephric/Wolffian duct abnormalities in the pediatric population’, Canadian Urological Association Journal, vol. 14, no. 11, p. E611, 2020. [2] S. Ruffenach, ‘Caspar Friedrich Wolff (1734-1794)’, Embryo Project Encyclopedia, 2009. [3] A. P. Houat et al., ‘Congenital anomalies of the upper urinary tract: a comprehensive review’, Radiographics, vol. 41, no. 2, pp. 462–486, 2021. [4] K. M. Schmidt-Ott et al., ‘Novel regulators of kidney development from the tips of the ureteric bud.’, Journal of the American Society of Nephrology: JASN, vol. 16, no. 7, pp. 1993–2002, 2005. [5] R. Westland, M. F. Schreuder, J. C. Ket, and J. A. van Wijk, ‘Unilateral renal agenesis: a systematic review on associated anomalies and renal injury’, Nephrology Dialysis Transplantation, vol. 28, no. 7, pp. 1844–1855, 2013. [6] A. Hindryckx and L. De Catte, ‘Prenatal diagnosis of congenital renal and urinary tract malformations’, Facts, views & vision in ObGyn, vol. 3, no. 3, p. 165, 2011. [7] M. Kumar, G. Kumar, K. Barwal, and P. Raina, ‘Right supernumerary kidney: a rare entity’, Urology Case Reports, vol. 23, p. 97, 2019. [8] G. N’Guessan and F. D. Stephens, ‘Supernumerary kidney’, J Urol, vol. 130, no. 4, pp. 649–653, Oct. 1983, doi: 10.1016/s0022-5347(17)51385-3. [9] H.-Y. Tsai, M.-H. Lee, H.-C. Chen, H.-C. Chen, and J.-Y. Guh, ‘Sagittally malrotated kidney: a case series of two patients’, Surgical and Radiologic Anatomy, vol. 37, pp. 551–553, 2015. [10] E. Chang, C. Santillan, and M. K. O’Boyle, ‘Blind-ending branch of a bifid ureter: multidetector CT imaging findings’, The British Journal of Radiology, vol. 84, no. 998, pp. e38–e40, 2011. [11] S. Moghul, S. Liyanage, S. Vijayananda, M. Tam, and I. Kapralos, ‘Bifid ureter with blind-ending branch: A rare anatomic variant detected during antegrade ureteric stent insertion’, Radiology Case Reports, vol. 13, no. 6, pp. 1199–1202, 2018. [12] T. Berrocal, P. López-Pereira, A. Arjonilla, and J. Gutiérrez, ‘Anomalies of the distal ureter, bladder, and urethra in children: embryologic, radiologic, and pathologic features’, Radiographics, vol. 22, no. 5, pp. 1139–1164, 2002, doi: 10.1148/radiographics.22.5.g02se101139. [13] P. L. Dwyer and A. Rosamilia, ‘Congenital urogenital anomalies that are associated with the persistence of Gartner’s duct: a review’, American journal of obstetrics and gynecology, vol. 195, no. 2, pp. 354–359, 2006. [14] O. Ocal, A. D. Karaosmanoglu, M. Karcaaltıncaba, D. Akata, and M. Ozmen, ‘Imaging findings of congenital anomalies of seminal vesicles’, Polish Journal of Radiology, vol. 84, pp. 25–31, 2019. [15] H. M. Shebel, H. M. Farg, O. Kolokythas, and T. El-Diasty, ‘Cysts of the lower male genitourinary tract: embryologic and anatomic considerations and differential diagnosis’, Radiographics, vol. 33, no. 4, pp. 1125–1143, 2013, doi: 10.1148/rg.334125129. [16] E. O. Pacheco et al., ‘Male pelvic cysts: a didactic diagnostic approach’, RadioGraphics, vol. 41, no. 6, pp. E179–E180, 2021. [17] M. Nistal, P. González-Peramato, G. Sousa, M. A. García-Cabezas, J. I. Rodríguez, and M. M. Cajaiba, ‘Cystic dysplasia of the epididymis: a disorder of mesonephric differentiation associated with renal maldevelopment’, Virchows Arch, vol. 456, no. 6, pp. 695–702, Jun. 2010, doi: 10.1007/s00428-010-0906-8. [18] S. Vohra and A. Morgentaler, ‘Congenital anomalies of the vas deferens, epididymis, and seminal vesicles’, Urology, vol. 49, no. 3, pp. 313–321, Mar. 1997, doi: 10.1016/S0090-4295(96)00433-5. [19] M. Valentino, M. Bertolotto, M. Ruggirello, P. Pavlica, L. Barozzi, and C. Rossi, ‘Cystic lesions and scrotal fluid collections in adults: Ultrasound findings’, J Ultrasound, vol. 14, no. 4, pp. 208–215, Nov. 2011, doi: 10.1016/j.jus.2011.10.008. [20] R. H. Donkol, ‘Imaging in male-factor obstructive infertility’, World Journal of Radiology, vol. 2, no. 5, p. 172, 2010. [21] P. K. Mittal et al., ‘Role of imaging in the evaluation of male infertility’, Radiographics, vol. 37, no. 3, pp. 837–854, 2017. [22] J. W. McQuaid and C. Tanrikut, ‘Ejaculatory Duct Obstruction: Current Diagnosis and Treatment’, Curr Urol Rep, vol. 14, no. 4, pp. 291–297, Aug. 2013, doi: 10.1007/s11934-013-0340-y. [23] B. E. Howitt and M. R. Nucci, ‘Mesonephric proliferations of the female genital tract’, Pathology, vol. 50, no. 2, pp. 141–150, Feb. 2018, doi: 10.1016/j.pathol.2017.11.084. [24] D. K. Walker, R. A. Salibian, A. D. Salibian, K. M. Belen, and S. L. Palmer, ‘Overlooked Diseases of the Vagina: A Directed Anatomic-Pathologic Approach for Imaging Assessment’, RadioGraphics, vol. 31, no. 6, pp. 1583–1598, Oct. 2011, doi: 10.1148/rg.316115531. [25] K. S. Eilber and S. Raz, ‘Benign cystic lesions of the vagina: a literature review’, The Journal of urology, vol. 170, no. 3, pp. 717–722, 2003. [26] P. L. Moyle, M. Y. Kataoka, A. Nakai, A. Takahata, C. Reinhold, and E. Sala, ‘Nonovarian cystic lesions of the pelvis’, Radiographics, vol. 30, no. 4, pp. 921–938, 2010. [27] A. Hofmann, F. Vauth, and W. H. Roesch, ‘Zinner syndrome and infertility─a literature review based on a clinical case’, Int J Impot Res, vol. 33, no. 2, Art. no. 2, Mar. 2021, doi: 10.1038/s41443-020-00360-0. [28] M. Campora et al., ‘Zinner Syndrome: A Diagnostic Challenge. The Aid of Morphology, Embryology, and Immunohistochemistry’, Urology, vol. 108, pp. e3–e5, Oct. 2017, doi: 10.1016/j.urology.2017.06.013. [29] J. M. Lee, ‘Herlyn-Werner-Wunderlich Syndrome: A Mini-review’, Child Kidney Dis, vol. 22, no. 1, pp. 12–16, Apr. 2018, doi: 10.3339/jkspn.2018.22.1.12. [30] P. Acién and M. I. Acién, ‘The history of female genital tract malformation classifications and proposal of an updated system†’, Human Reproduction Update, vol. 17, no. 5, pp. 693–705, Sep. 2011, doi: 10.1093/humupd/dmr021. [31] L. Zhu, N. Chen, J.-L. Tong, W. Wang, L. Zhang, and J.-H. Lang, ‘New Classification of Herlyn-Werner-Wunderlich Syndrome’, Chin Med J (Engl), vol. 128, no. 2, pp. 222–225, Jan. 2015, doi: 10.4103/0366-6999.149208. [32] G. F. Grimbizis et al., ‘The ESHRE/ESGE consensus on the classification of female genital tract congenital anomalies’, Hum Reprod, vol. 28, no. 8, pp. 2032–2044, Aug. 2013, doi: 10.1093/humrep/det098. [33] R. Coleman et al., ‘Tubulocystic anomalies of the mesonephric duct associated with ipsilateral renal dysgenesis’, J Pediatr Urol, vol. 15, no. 1, p. 46.e1-46.e6, Feb. 2019, doi: 10.1016/j.jpurol.2018.07.021. [34] J. Pors et al., ‘Clinicopathologic characteristics of mesonephric adenocarcinomas and mesonephric-like adenocarcinomas in the gynecologic tract: a multi-institutional study’, The American journal of surgical pathology, vol. 45, no. 4, p. 498, 2021. [35] S. Stolnicu et al., ‘International Endocervical Adenocarcinoma Criteria and Classification (IECC): a new pathogenetic classification for invasive adenocarcinomas of the endocervix’, The American journal of surgical pathology, vol. 42, no. 2, p. 214, 2018. [36] H. Lee, H. Kim, and H.-S. Kim, ‘Mesonephric adenocarcinoma of the vagina harboring TP53 mutation’, Diagnostics, vol. 12, no. 1, p. 119, 2022. [37] C. Xie, Q. Chen, and Y. Shen, ‘Mesonephric adenocarcinomas in female genital tract: A case series’, Medicine, vol. 100, no. 35, 2021. [38] A. Dierickx, M. Göker, G. Braems, P. Tummers, and R. Van den Broecke, ‘Mesonephric adenocarcinoma of the cervix: case report and literature review’, Gynecologic Oncology Reports, vol. 17, pp. 7–11, 2016. [39] K. Seay, T. Akanbi, B. Bustamante, S. Chaudhary, and G. L. Goldberg, ‘Mesonephric-like adenocarcinoma of the ovary with co-existent endometriosis: A case report and review of the literature’, Gynecol Oncol Rep, vol. 34, p. 100657, Oct. 2020, doi: 10.1016/j.gore.2020.100657.